taxonomer.iobio
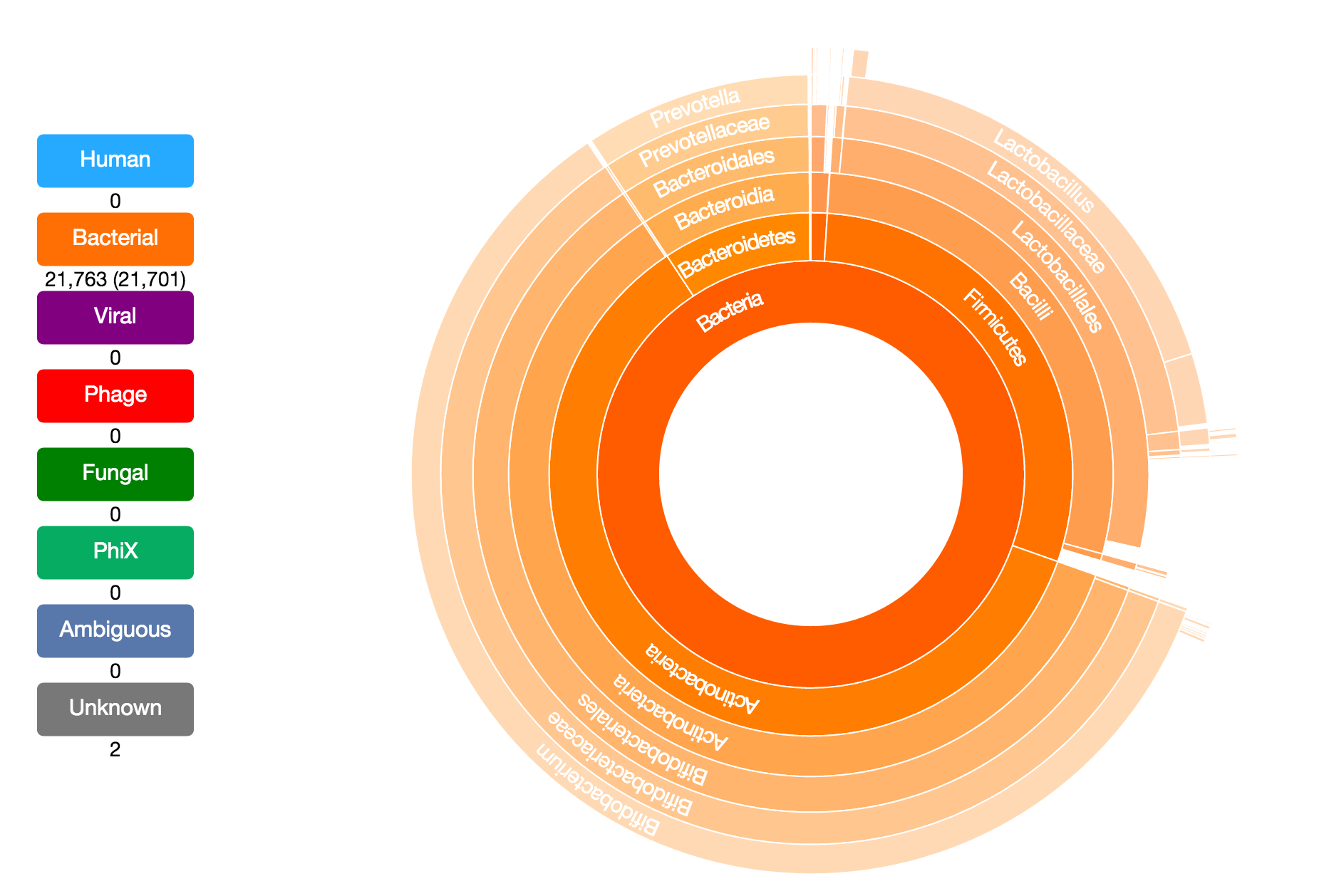
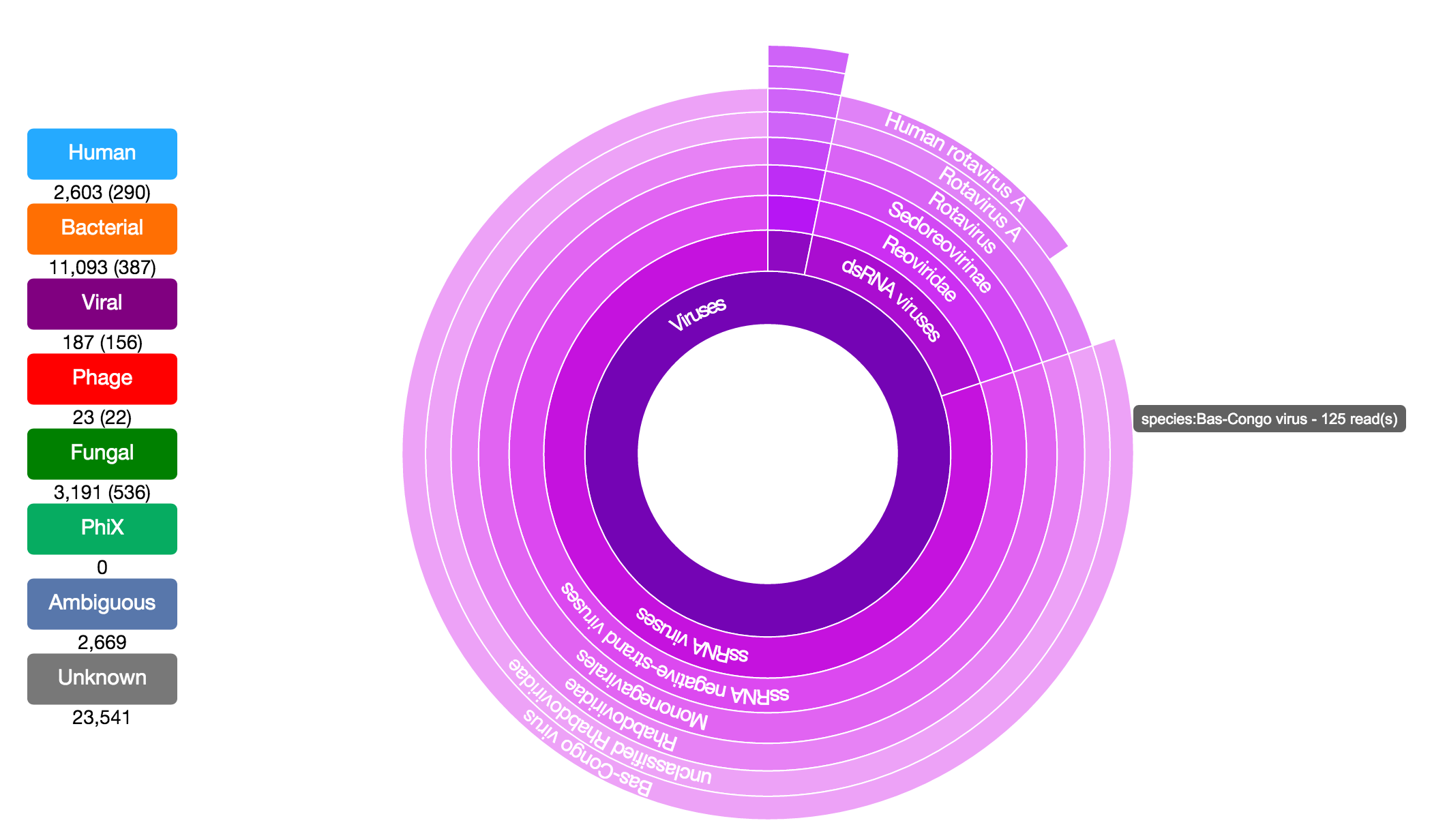
Taxonomer is a kmer-based ultrafast metagenomics tool for assigning taxonomy to sequencing reads from both clinical and environmental samples. Taxonomer enables simultaneous microbial detection and human transcript quantification within a clinically meaningful timeframe (i.e. minutes). Taxonomer supports nucleotide and protein-based pathogen detection and phylogenetic classification using DNA and RNA sequencing data in a single integrated algorithmic framework.
Taxonomer results are interactively visualized through a real-time web service on the
iobio visualization toolkit at
taxonomer.iobio. This allows rapid, highly interactive analyses using personal computers and mobile devices. This app enables scientists and clinicians to examine the composition of their metagenomic samples in seconds and further explore the taxonomic classifications interactively without the overhead of bioinformatics expertise.
Users can explore the demo datasets described below or analyze their own data (FASTQ or FASTA format sequence files). The user has the option to either select a file on an http accessible site or on their hard drive. For demo purposes, the app will read and analyze the first few thousand reads from the file.
Taxonomer was developed in collaboration between 3 laboratories at the University of Utah: the Schlaberg lab in the Department of Pathology and at ARUP Laboratories, the Eilbeck lab in the Department of Biomedical Informatics, and the Yandell lab in the Department of Human Genetics. Graduate students Steven Flygare (Mark Yandell Lab) and Keith Simmon (Karen Eilbeck's Lab) developed the algorithms and computational methods of taxonomer. Steven developed the statistical algorithms and wrote and optimized taxonomer's codebase. Keith Simmon organized and curated microbiological and sequence data sources to create taxonomer backend databases and created in depth benchmarks to extensively validate taxonomer.The Taxonomer.iobio web app is built upon the Marth lab iobio platform, implemented by Chase Miller, Yi Qiao and Tonya Di Sera.
Taxonomer development was supported by the Utah Science Technology and Research (USTAR) Center for Genetic Discovery; the Primary Children’s Hospital Foundation; the University of Utah’s Center for Clinical and Translational Science (CCTS), the Richard A. and Carol M. Fay Endowed Graduate Fellowship, the Department of Pathology and NIH training grants 1ULTR001067 and 5T32HL007576-29. The iobio platform is supported by grant U01 HG006513 to Gabor Marth from the National Human Genome Research Institute.
IDbyDNA has licensed the Taxonomer technology from the University of Utah, further developed the Taxonomer software, and created more comprehensive classification databases. As part of a collaboration with
Frameshift Genomics, it has taken the visualization features of the free academic site, and added further iobio-powered user interface features. The resulting “freemium” analysis app expands the search to include bacterial and viral Uniref sequences, and its Full Analysis mode allows offline, but complete, analysis of a dataset with even tens of millions of sequencing reads. Try this tool at
taxonomer.com
Demo Datasets